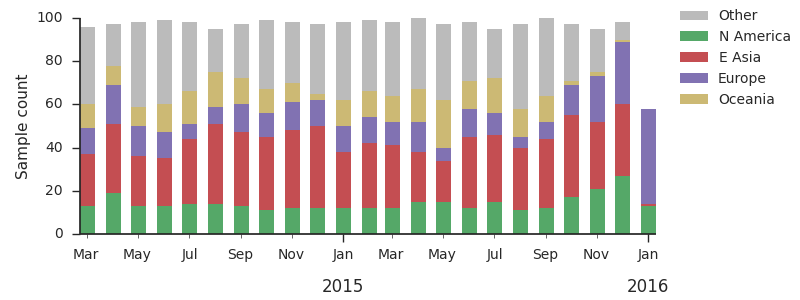
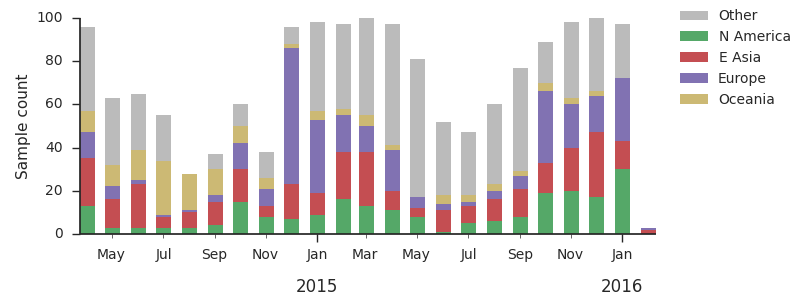
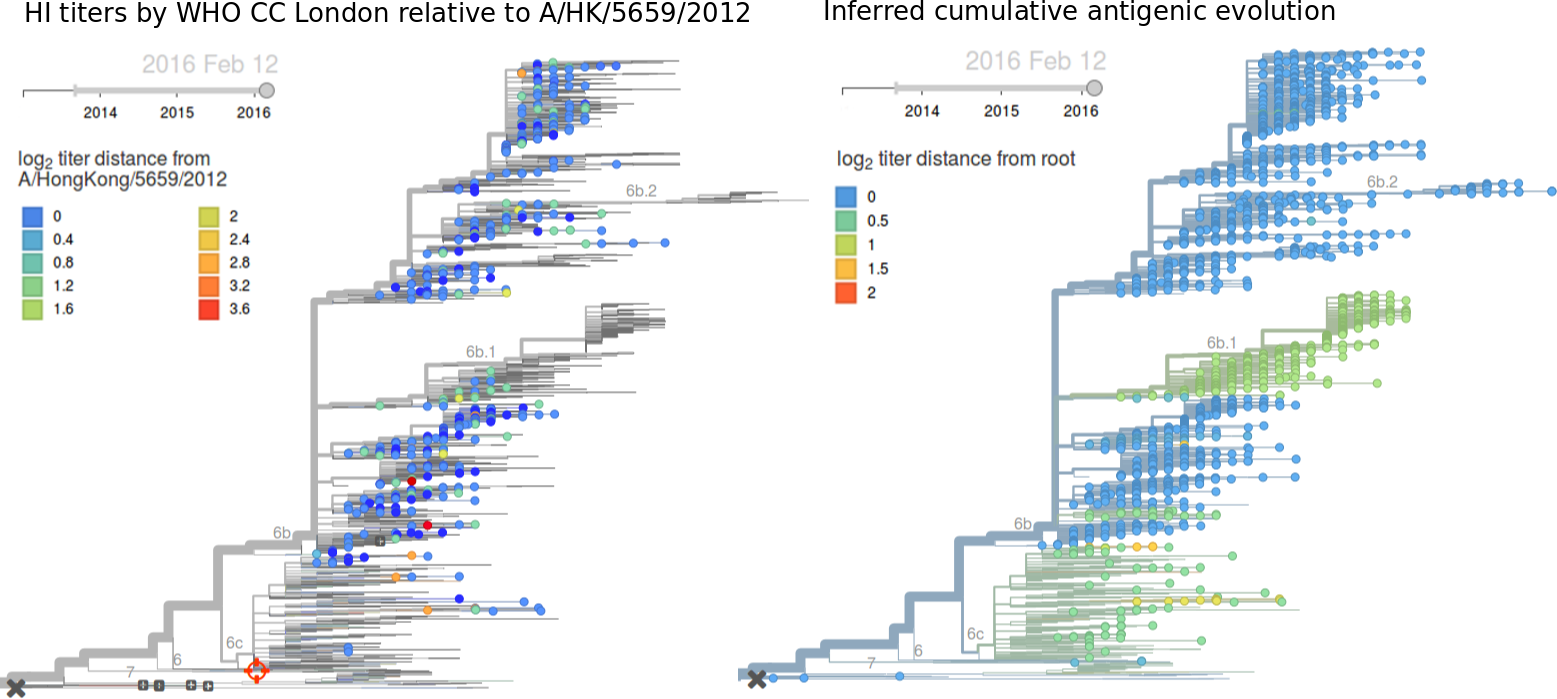
For each virus, we know its date of collection and we know its genetic make up, allowing us count the number of viruses belonging to a particular genetic lineage through time. We use these timeseries of presence/absence to estimate clade frequencies through time. This estimation smooths month-to-month sampling variation.
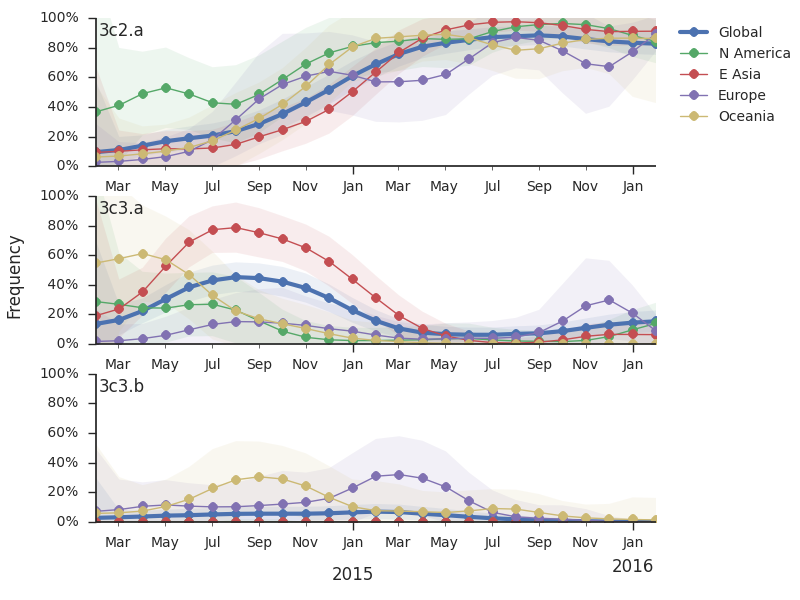
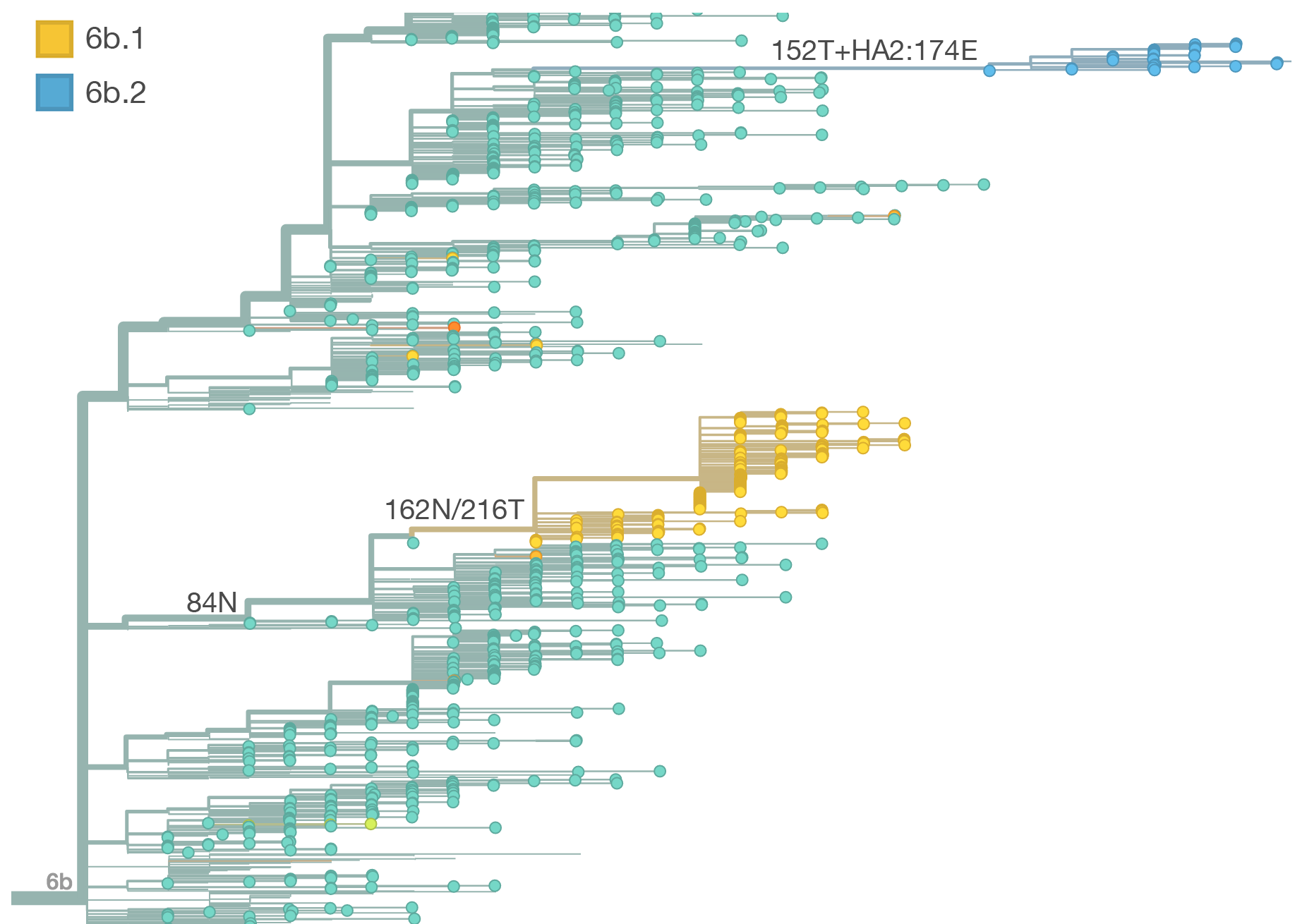
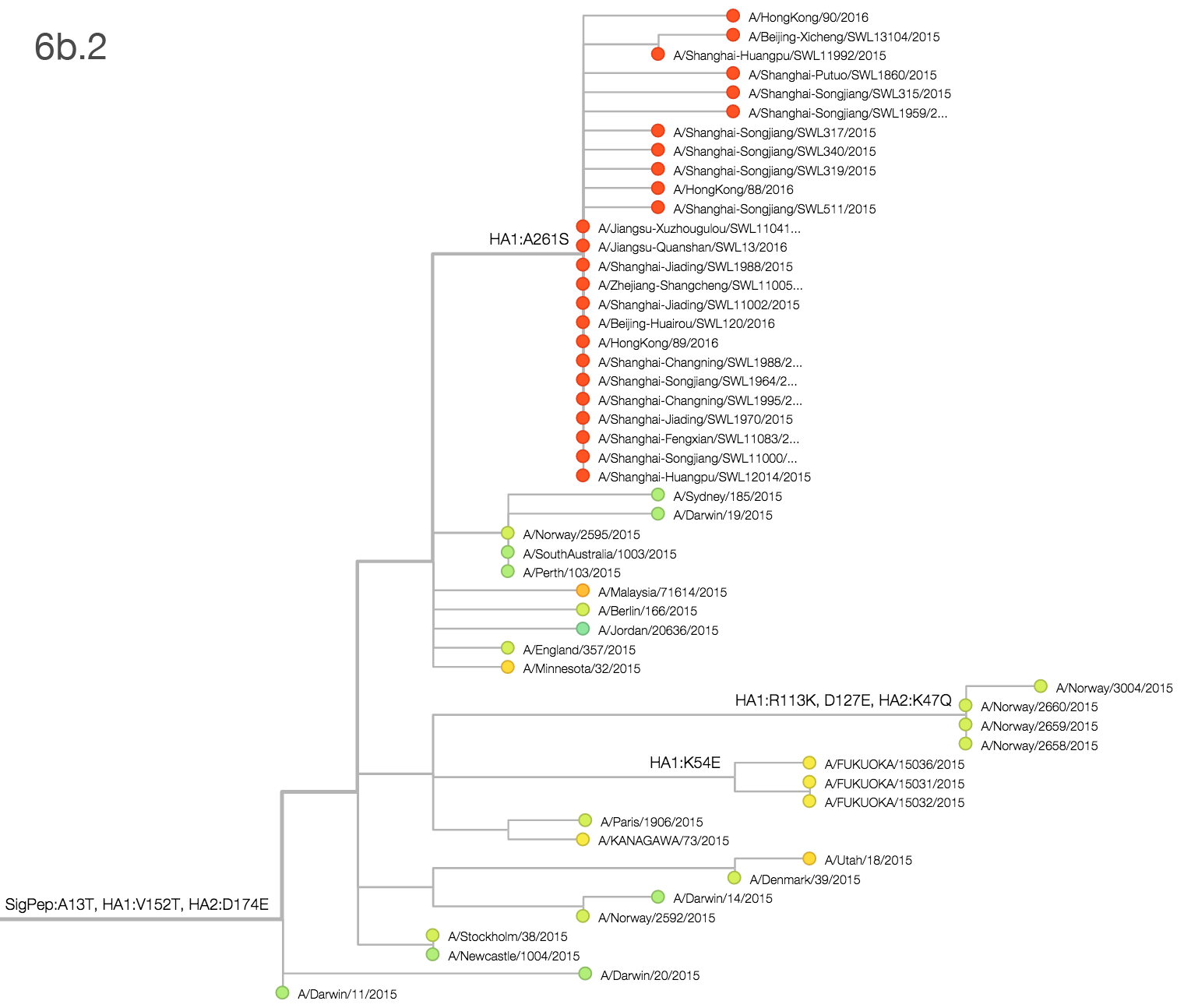
Throughout 2015, clade 3c2.a viruses predominated over 3c3.a and 3c3.b viruses. The only exception to this trend has been the recent reemergence of 3c3.a viruses in Europe. However, this clade of European 3c3.a viruses lacks recent amino acid substitutions relative to the 3c3.a parent. Thus it appears that the reemergence of 3c3.a viruses in Europe is due to epidemiological factors, rather than genetic factors. In addition, we find that 3c2.a viruses still predominate in Europe over 3c3.a viruses despite their recent uptick and 3c3.a isolates have been declining in recent weeks.

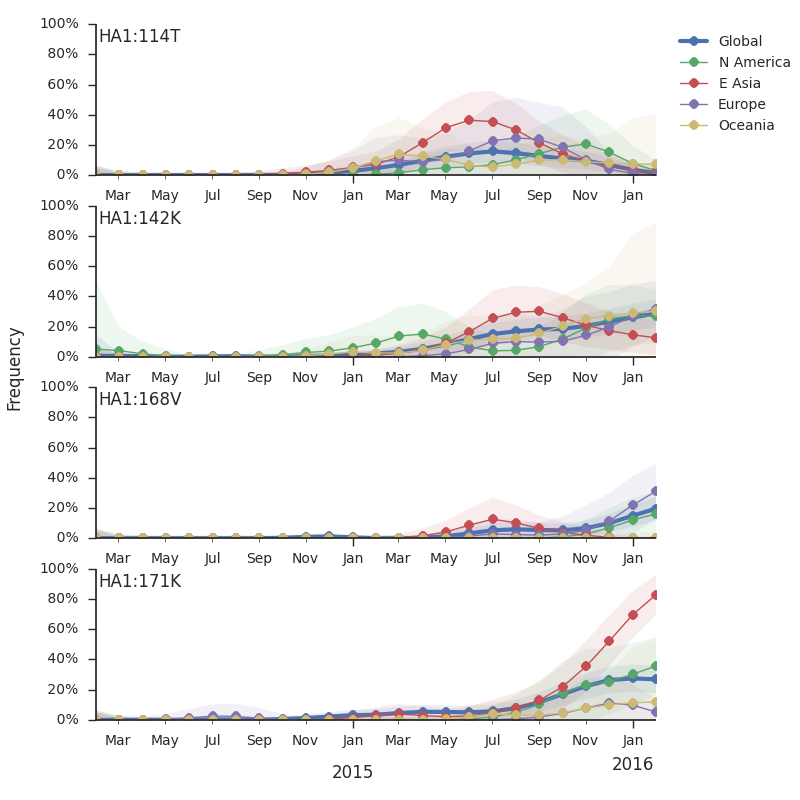
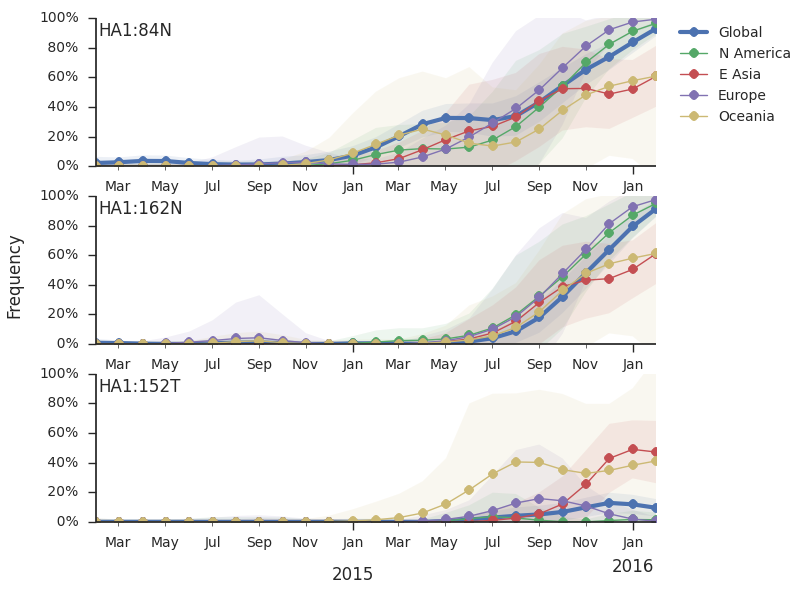
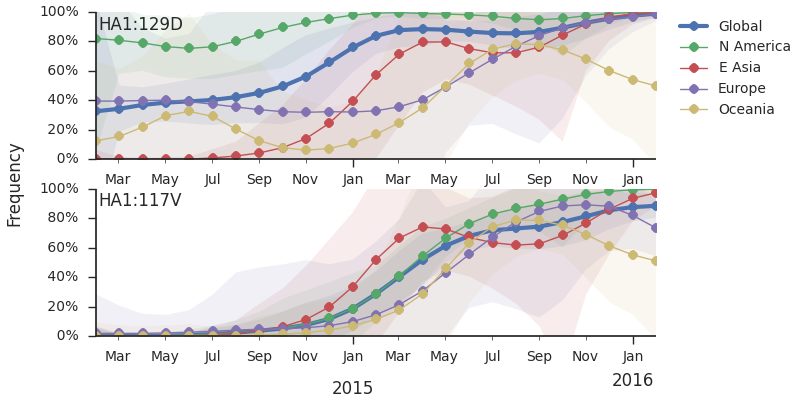
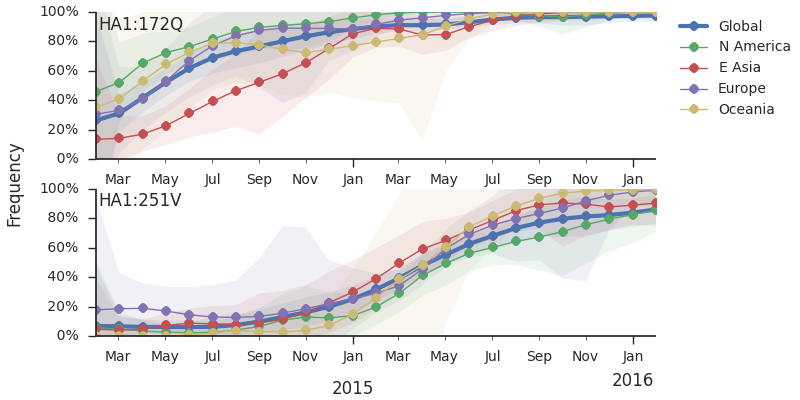
The 114T variant peaked in frequency around July 2015 and similarly the 142K variant has been around for more than a year with a low but even geographic distribution, although has seen a recent uptick in frequency since October due to 168V. We expect neither of these variants to be under strong positive selection. The T160K substitution is observed scattered within clade 3c2.a and overrepresented in sequences obtained from cultured virus.
We observe a recent rapid increase in frequency of (HA1:171K, HA2:77V/155E) especially in Asia and North America. This has been the fastest growing variant within 3c2.a that we have observed. This clade also shows rapid phylogenetic branching, corroborating its rapid rise. Additionally, the subclade bearing HA1:142K/197R/168V has recently risen in frequency, although not as rapidly as the (HA1:171K, HA2:77V/155E) clade. We estimate clade HA1:142K/197R/168V has gone from 1% global frequency in March 2015 to 19% global frequency today, while clade (HA1:171K, HA2:77V/155E) has gone from 1% global frequency in June 2015 to 29% global frequency today. For comparison, 3c3.a viruses increased in frequency from 1% to 31% in a similar span of 8 months time.
Barring substantial changes in other clades, we predict the (HA1:171K, HA2:77V/155E) variant to dominate.
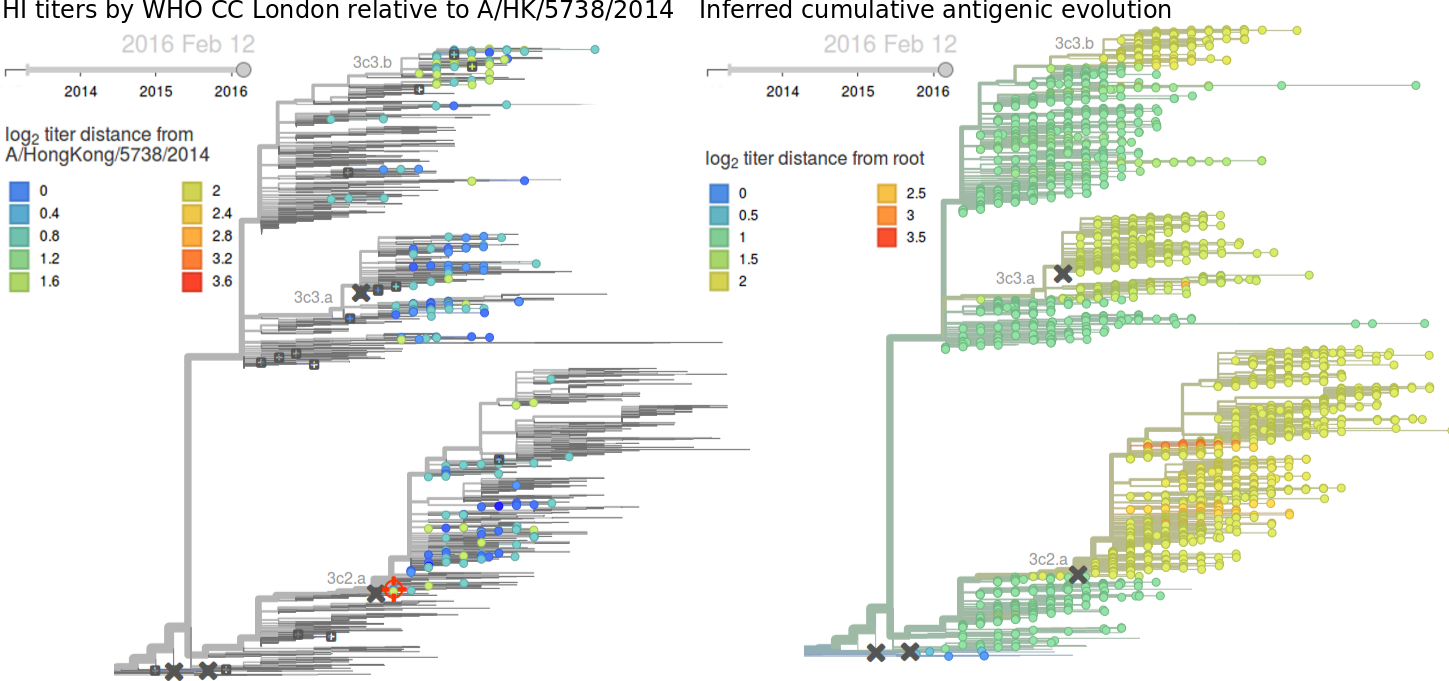


Written by Trevor Bedford and Richard Neher. This work is made possible by the GISAID Initiative and the open sharing of genetic data by influenza research groups from all over the world. We gratefully acknowledge their contributions. Give us a shout at @trvrb or @richardneher with questions or comments.